 v.5.0 build 2020-09
v.5.0 build 2020-09
About FunCoup
The name FunCoup [fən kəp] stands for functional coupling. FunCoup is a framework to infer genome-wide functional couplings in 21 model organisms. Functional coupling, or functional association, is an unspecific form of association that encompasses direct physical interaction but also more general types of direct or indirect interaction like regulatory interaction or participation in the same process or pathway.
Framework
Briefly, the FunCoup framework integrates 10 different evidence types derived from high-throughput genomics and proteomics data in a naive Bayesian integration procedure. The evidence types are discussed in more detail below. Evidence is transferred across species using orthology assignments from InParanoid.
The naive Bayesian integration combines the likelihood for coupling and no coupling in the form of log-likelihood ratios (LLRs) for all data sets. LLRs for data of the same type are corrected to account for cross-data redundancies. The sum of LLRs for a gene pair is called the final Bayesian score (FBS) and expresses the amount of support the data shows for a coupling. To simplify the interpretation the FBS is transformed into a probabilistic confidence score that ranges from 0 to 1. For more details, please have a look at the FunCoup publications.
Networks
FunCoup differentiates between five different classes of functional couplings: protein-protein interaction (PPI), complex co-membership, co-membership in a metabolic pathway, co-membership in a signaling pathway and shared operon. For each class a separate network is created. Additionally a composite or summary network is created by taking the strongest coupling from the different classes for each pair.
Tutorials
A playlist of video tutorials with an introduction and demos on FunCoup can be found on YouTube.
Additional resources, e.g. slides and an R-notebook on offline usage, can be found in Google Drive.
Evidence types
Evidences are the signals that support or contradict the presence of functional coupling. Typically some kind of scoring function is used to convert raw data into evidence. For a complete list of all data see here. FunCoup integrates 9 different evidence types listed below.
Protein interaction (PIN)
Physical protein interactions (PINs) from iRefIndex are combined, where interactions confirmed by multiple publications get a higher score. The scoring function further down-weights interactions from large scale experiments and prey-prey interactions.
mRNA co-expression (MEX)
mRNA co-expression across multiple experimental conditions or tissues provides a strong signal for functional coupling. FunCoup evaluates co-expression as Spearman correlation of expression profiles. For each species multiple selected large scale experiments from GEO are integrated.
Protein co-expression (PEX)
The concordance between mRNA and protein expression is low. Directly measured protein expression from the Human protein atlas provides a more accurate estimation of protein abundances and is used to complement the mRNA expression data.
Genetic interaction profile similarity (GIN)
FunCoup does not explicitly consider genetic interactions as functional coupling. Rather, between pathway genetic interactions are integrated in the form of genetic interaction profile similarity. The underlying assumption is that genes in the same process or pathway have similar genetic interactions with genes in other alternative processes or pathways.
Shared transcription factor binding (TFB)
Genes are regulated by multiple transcription factors (TFs) and FunCoup uses TF profile similarity as evidence for functional coupling.
Co-miRNA regulation by shared miRNA targeting (MIR)
Similar to shared transcription factor binding, co-regulation by multiple miRNA is used as evidence for function coupling.
Sub-cellular co-localization (SCL)
Shared sub-cellular localization and dissimilar localization are good positive and negative indicators for functional couplings. FunCoup uses localizations from the cellular component GO ontology. Co-localizations are weighted by the specificity of the localization, where specific localizations get a high weight and unspecific localizations get a low weight.
Domain interactions (DOM)
Predicted domain interactions from UniDomInt are used as evidence. The confidence score provided by UniDomInt is summed up for all domain pairs of two proteins.
Phylogenetic profile similarity (PHP)
A phylogenetic profile is a gene conservation pattern across multiple species. Phylogenetic profile similarity provides an indication for functional coupling. FunCoup scores profile similarity as a fraction of branch lengths shared by both genes or exclusively covered by only one gene in a phylogeny of 273 species derived from InParanoid.
Quantitative mass spectrometry (QMS)
QMS data sets were obtained via PaxDB(v. 4.0). In a preprocessing step only the highest abundant proteins per condition were extracted and labeled accordingly. These profiles were further evaluated using an adapted Jaccard index (12). Here two proteins being abundant across different tissues would achieve high similarity scores.
Gene Regulatory (GRG)
Directed links inferred from transcription factor-gene bindings. Gold standards for this network type are obtained from TRRUSTv2 and RegNetwork ChIP-seq data from ENCODE is used as evidence, where links are assigned probabilistic scores of regulation via Target Identification from Profiles (TIP).
Gold Standards
PPI
Gold standard couplings for PPI (Protein-protein interactions) were collected from iRefIndex (v16). This gold standard source is intersected with gold standards Complex, Metabolic and Signaling, so ensure high reliability in the protein interactions.
Complex
Gold standard couplings for protein complexes were collected from iRefIndex (v16), ComplexPortal (2020-05) and Corum (v3). The protein complexes from iRefIndex were filtered to exclude very large complexes including large parts of the proteome.
Metabolic
Metabolic gold standard couplings were collected from metabolic pathways obtained from KEGG (v94.1).
Signaling
Signaling gold standard couplings were collected from signaling pathways obtained from KEGG (v94.1).
Operons
Gold standard couplings from shared Operons were collected from OperonDB (v4). The operon gold standard is available for e.coli, c.elegans and b.subtilis.
Citation
Persson, E., Castresana-Aguirre, M., Buzzao, D., Guala, D., Sonnhammer, E, L. (2021)
FunCoup 5: Functional Association Networks in All Domains of Life, Supporting Directed Links and Tissue-Specificity
Journal of molecular biology, 433, 166835.
Ogris, C., Guala, D., Kaduk, M., Sonnhammer, E. L. (2017)
FunCoup 4: new species, data, and visualization.
Nucleic Acids Research 46 (Database issue), D601-D607.
Schmitt, T., Ogris, C., & Sonnhammer, E. L. (2013).
FunCoup 3.0: database of genome-wide functional coupling networks.
Nucleic Acids Research, 42(Database issue), D380-8
Alexeyenko, A., Schmitt, T., Tjärnberg, A., Guala, D., Frings, O., & Sonnhammer, E. L. (2012).
Comparative interactomics with Funcoup 2.0.
Nucleic Acids Research, 40(Database issue), D821-8
Alexeyenko, A., & Sonnhammer, E. L. (2009).
Global networks of functional coupling in eukaryotes from comprehensive data integration.
Genome Research, 19(6), 1107-1116
License

This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Search options
Normal search
The default query retrieves the most strongly connected genes to one or multiple genes from the selected species network. The query searches for exact matches of symbols or identifiers and supports a variety of different identifier types including Ensembl gene, protein, and transcript IDs, NCBI gene IDs ,RefSeq IDs and UniProt IDs. For a search with multiple genes the identifiers should be separated by spaces. To get more control and alternative query options expand the advanced search options.
SARS_CoV-2 search
To simplify searching in the virus-host network for SARS-CoV-2 and Homo sapiens, we have introduced a specific button just for this purpose. The button can be found beneath the regular search-button, and clicking it will pre-fill a query with the SARS-CoV-2 genes, a confidence cutoff of 0.3, 50 nodes per expansion step, and then perform the search.
Advanced search
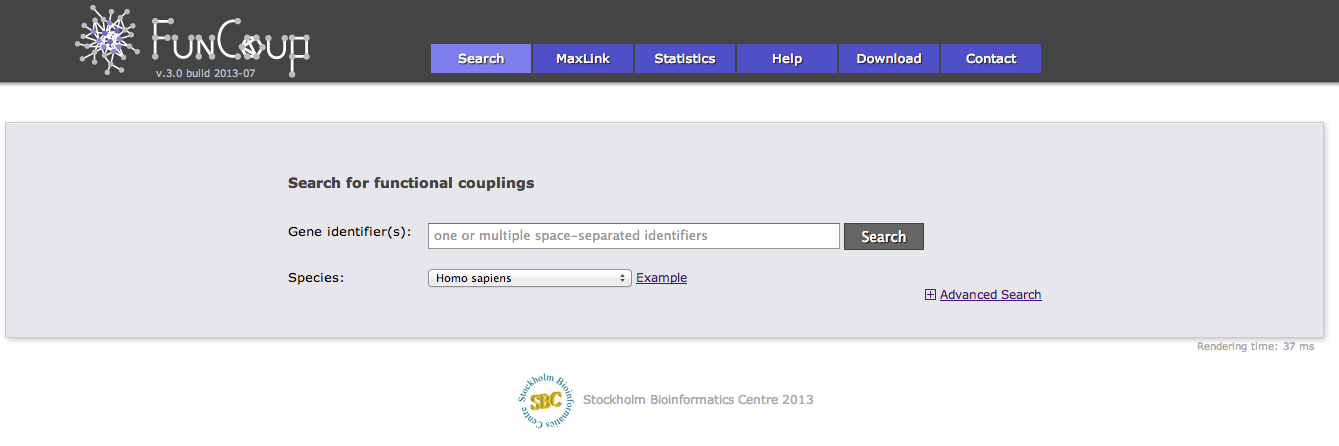
There are 4 different categories of advanced search options. The first category "Sub-network selection" controls how the subnetwork around the query is retrieved. The sub-network retrieval starts from the query genes and adds the top most strongly connected genes which have at least one connection to the query that is stronger than the given confidence threshold, finally all links between the retrieved gene set that are stronger than the threshold are added. Three parameters can be adjusted for this expansion: the confidence threshold, the number of most strongly connected genes that should be added, and how many expansion steps should be performed. If more than one expansion step (the default) is used, genes that were retrieved in the previous iteration are used as a query set in the next iteration and the process is repeated. If 0 expansion steps are selected only links between the query genes are retrieved and no genes are added.
There are 3 different algorithms to expand the network that differ in how multiple query genes are handled. The simplest algorithm retrieves the X strongest interactors to any of the query genes. If common neighbors are prioritized, all links to all query genes are considered and genes that are most strongly linked to many query genes are prioritized. Otherwise only the strongest link to a query gene counts. The third option is to treat the query genes as independent and retrieve the X interactors for every query gene.
The focus of FunCoup is the prediction of novel couplings, but known coupled genes that are part of the PPI or Complex gold standard can be added.
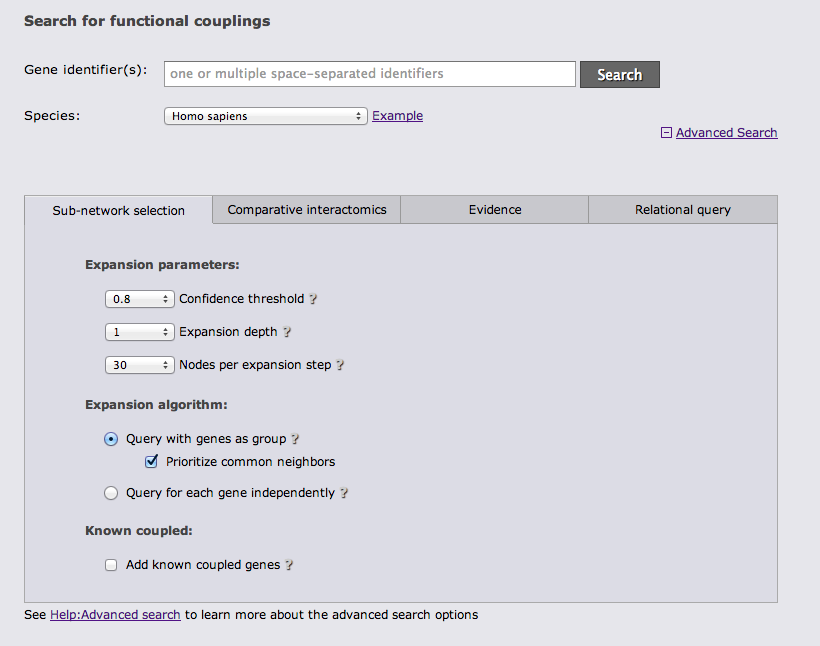
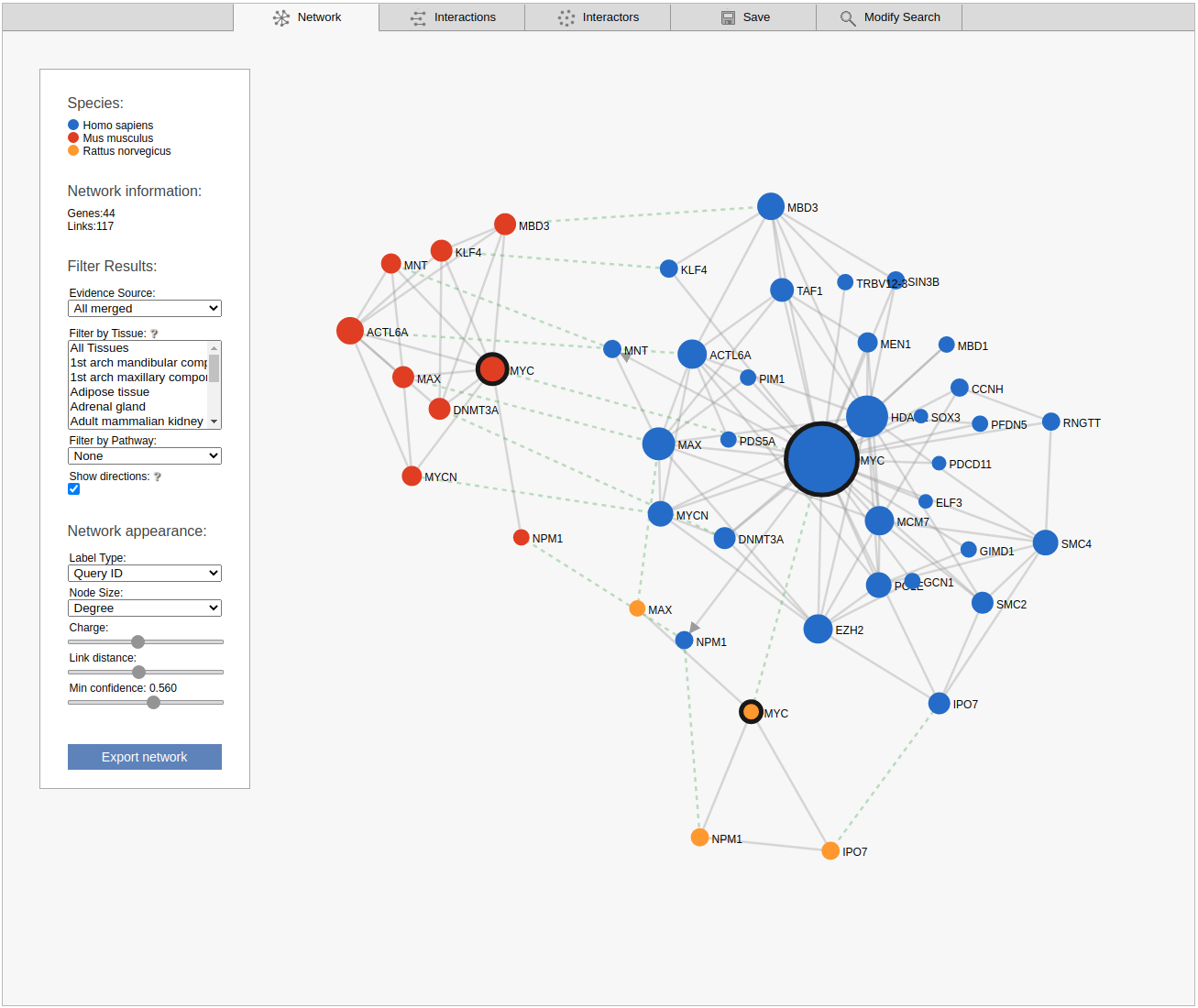
The next advanced search option tab allows you to run a comparative query across the networks of multiple species. This query retrieves the orthologs to the query genes and the sub-networks around them that maximizes the number of conserved links. If the checkbox at the bottom of the tab is checked, the search requires sufficient species-specific evidence (determined by the automatically lowered threshold in the sub-network tab). If this option is not checked orthology transfer might led to spurious sub-networks conservation.
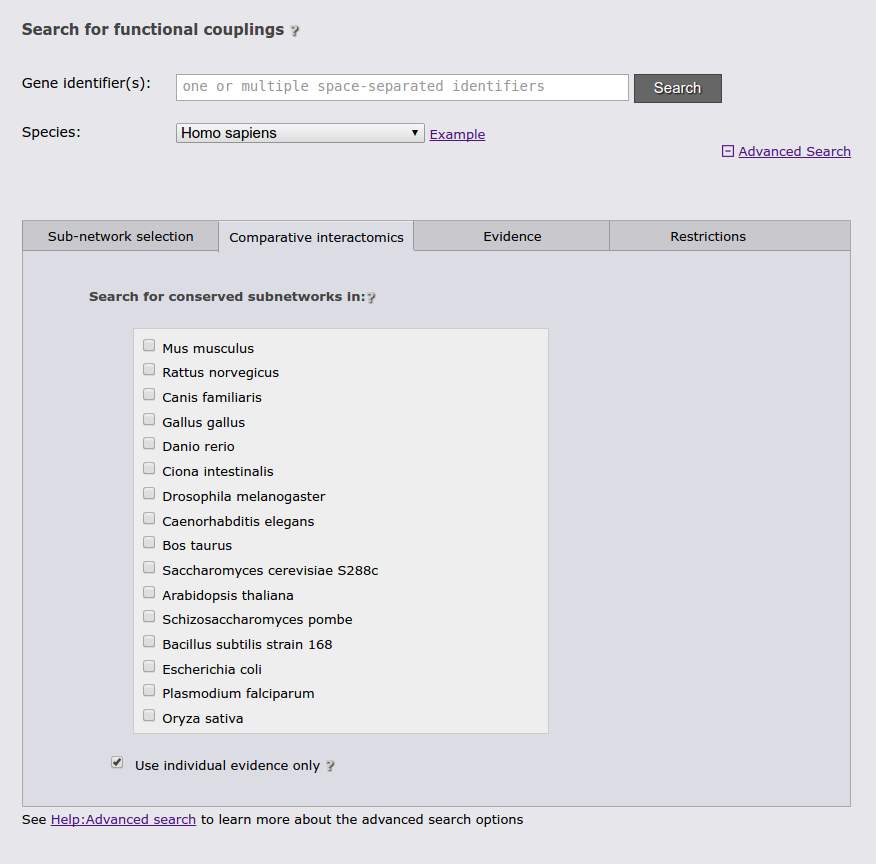
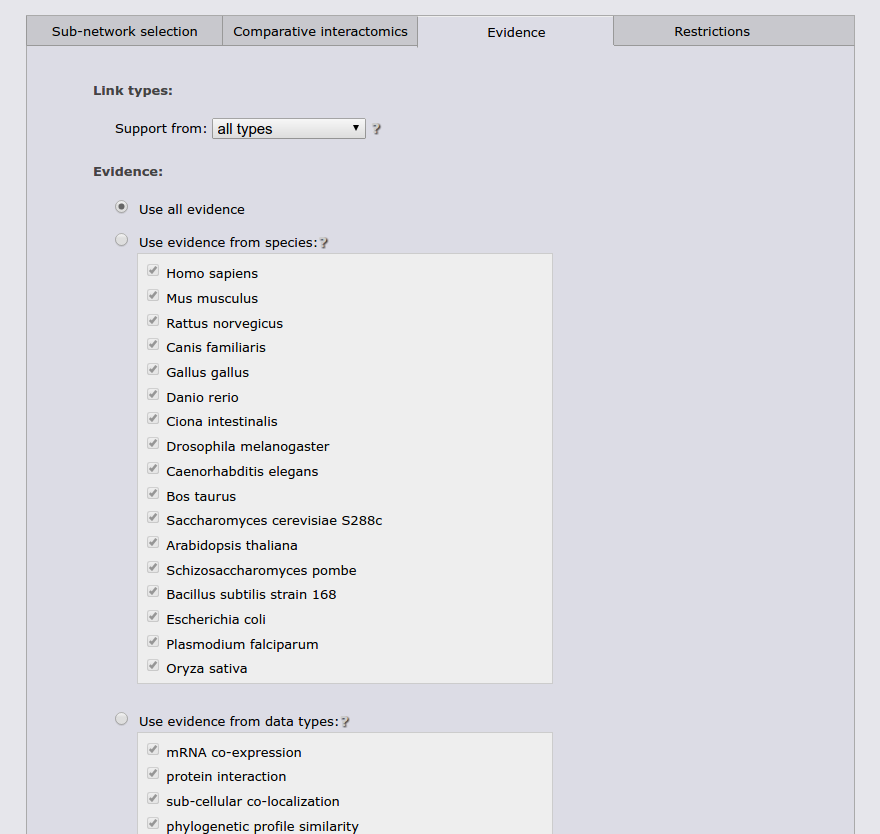
The next tab allows to restrict the search to a specific functional coupling class, by default the search operates on the strongest coupling class for each link. Furthermore, it is possible to require sufficient evidence from a subset of the evidence types or from a subset of species. It should however be noted that the display sub-network will always show all classes, species, and evidence types.
The last advanced search option tab allows to restrict the gene set from which the subnetwork is drawn either to a user-defined set of genes or to genes with a given annotation.
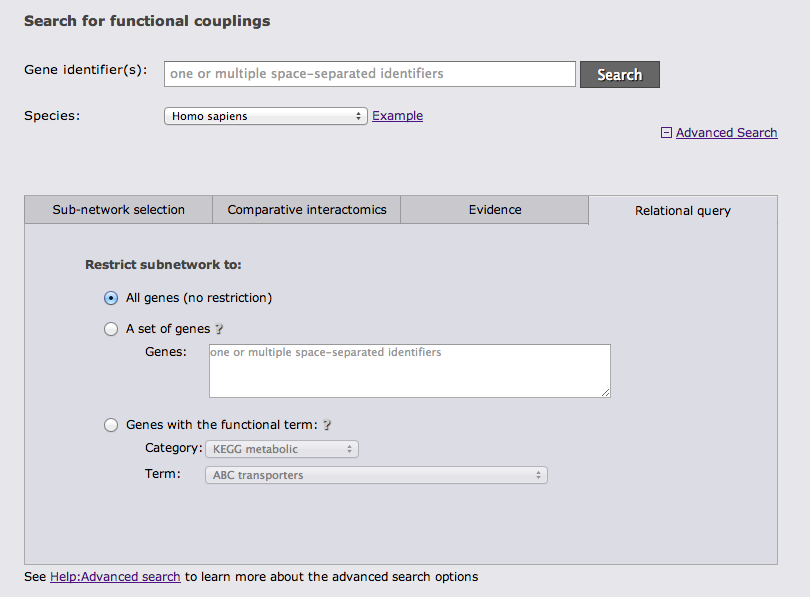
It is possible to combine search options from different tabs whenever this is sensible.
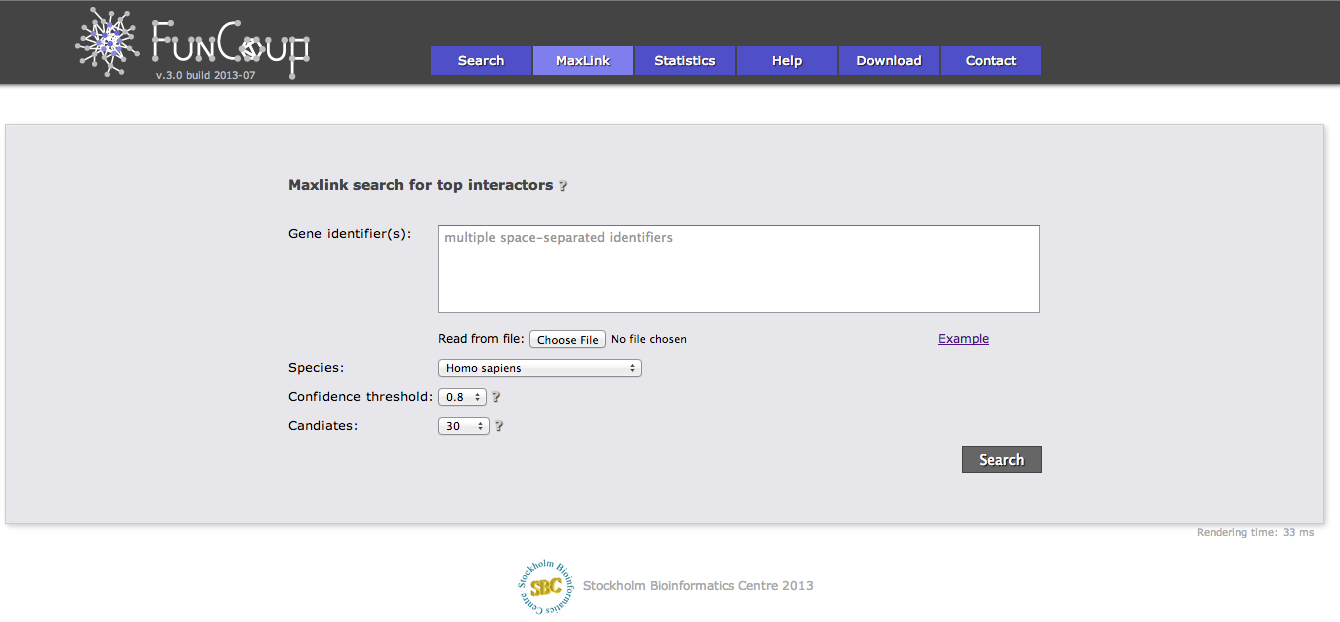
MaxLink search
The MaxLink search provides an alternative to the standard search. MaxLink has been successfully applied to predict novel cancer genes and was first described in Network-based Identification of Novel Cancer Genes (Östlund 2010). It is meant to be used with a long list of related query genes and retrieves genes that are signifcantly stronger connected to the query than expected by chance.
Result views
Network
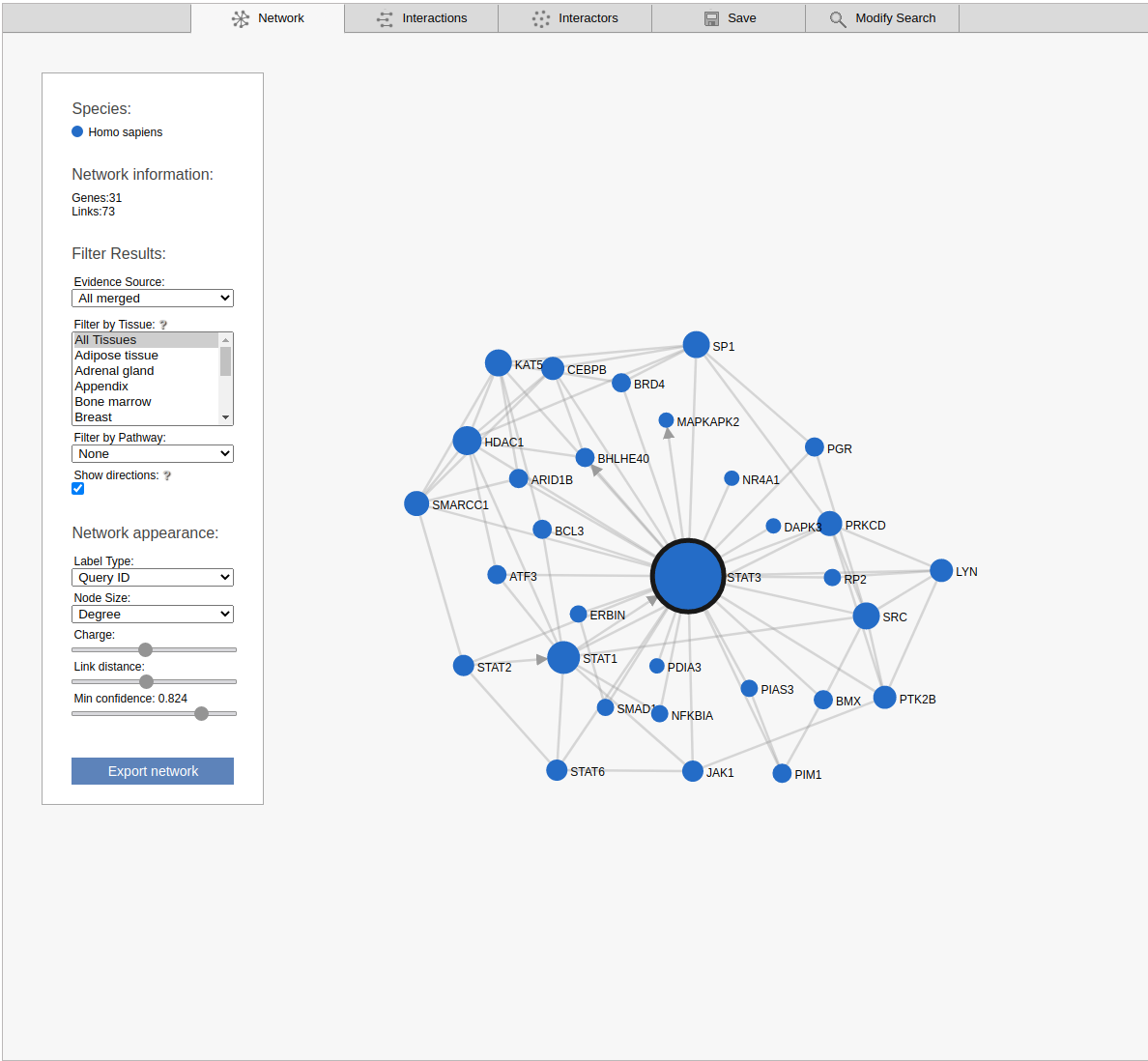
The network view displays the retrieved sub-network as a graph. Please note that the displayed network includes only the strongest links between the non-query subnetwork genes.
By default the viewer shows a summary network with the links from the strongest coupling class for each gene pair. The menu box on the left is grouped in four sections; Species, Network Information, Filter results and Network appearance. The sections Filter results and Network appearance have various options to manipulate the network. The Species section displays the name of the species included in your search, as well as the color of that species nodes. The Network information section displays additional information about a node or a link when the user hovers over it, otherwise the total number of genes and links within the subnetwork are shown. Within the Network appearance section the user can vary node Label and node Size, manipulate a node Charge, link distance and min confidence. Label: the default node label refers to the query identifier, but can be set to UniProt, Ensembl or NCBI ID. Additionally the label can also display species name, node degree or, if set to none, hide all the labels. Size: Node sizes scale with node degrees to emphasize gene importance. This can be adapted to scale depending on the number of participating pathways or not scale at all if set to none. Charge: This slider alters the tension between the nodes. The Filter results section contains four options, Evidence source, Filter by tissue, Filter by pathway and show directions. Evidence source: By default, a link represents the functional association inferred using all gold standards. Filter by tissue: Select one or multiple tissues to filter the subnetworks. By default, no tissue is selected Filter by pathway: This option is disabled by default. If a pathway is chosen the viewer highlights participating nodes in the species color, and the nodes not included in the pathway turns gray. Show directions: This option is only displayed when directed links are available. If at least one link in the subnetwork has a direction, the show directions checkbox will be selected by default. Uncheck the checkbox to hide the directions for the links. The Export network button allows downloading the subnetwork in the network viewer in .png format.
Interactions
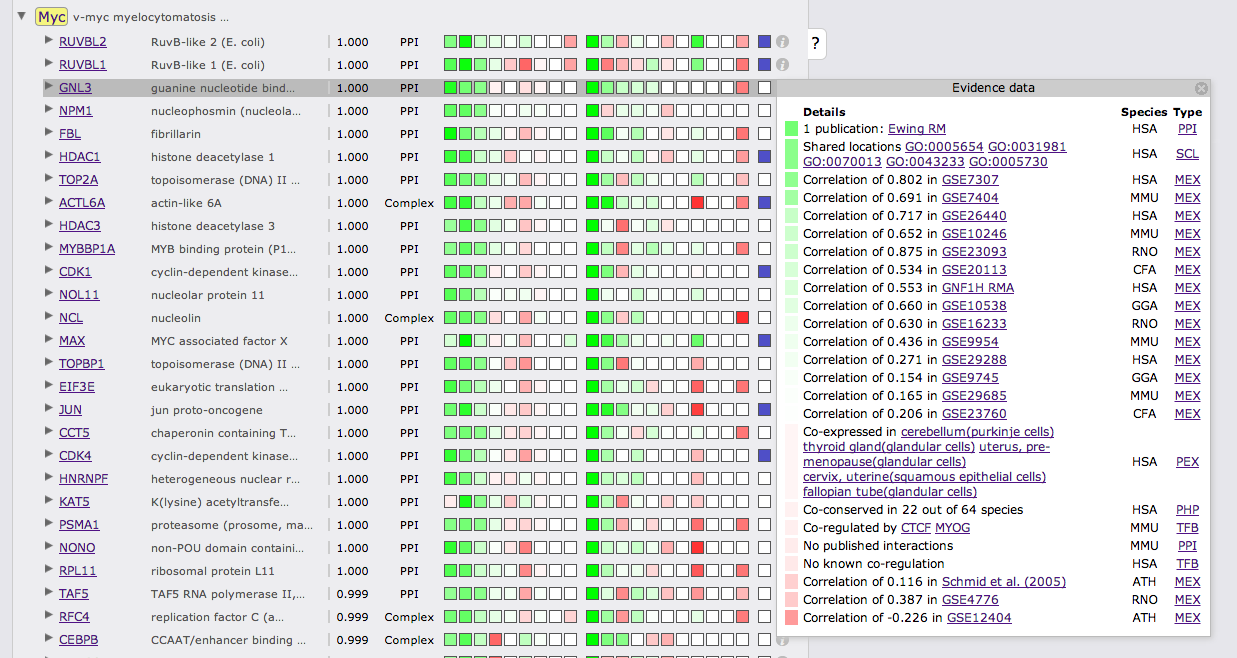
The interactions view lists all interactions between subnetwork genes and shows details about how the links that have been derived. The query genes are highlighted in yellow, query orthologs are highlighted in green. Clicking on a Gene identifier will bring up a box that allows you to use the gene or the pair as a query or to add the gene to the current query. Furthermore, cross-references and gene description are given. The green and red boxes represent positive and negative LLR for the different evidence types and species, hovering with the cursor over the box will display the LLR. Kown coupled pairs in the PPI or Complex gold standard are highlighted with a blue box. Initially only the strongest coupling class for each pair is shown. Clicking on the little triangle in front of the interaction partner will expand all other coupling classes.

Clicking on the green or red boxes or on the info symbol on the right will bring up a box displaying all evidence that led to the prediction. A green or red box shows if the evidence is positive or negative, hovering over the box will display the LLR. Next to the box there is a description of the evidence with cross links to data sources. The following two columns show the species from which the evidence stems and the type of the evidence.
Interactors
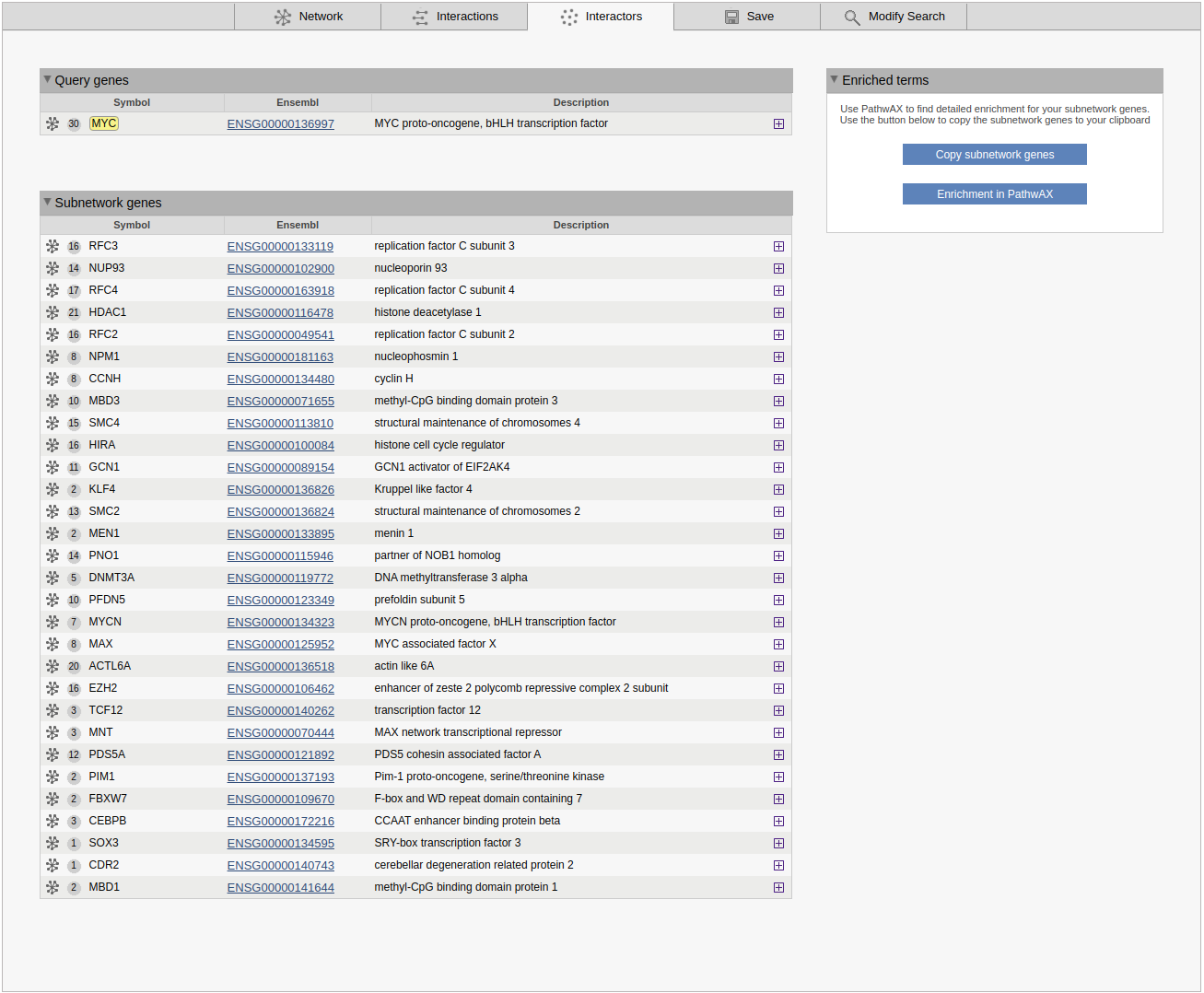
The interactors view gives an overview of all subnetwork genes. Query genes and other subnetwork genes are displayed in separete boxes. For each gene the symbol/displayed identifier and the Ensembl gene ID are shown. A grey circle in front of the identifier shows the degree of the gene in the sub-network clicking on the circle will highlight all connected genes. A network symbol to the left of the circle allows you to use the gene as a query or to add it to the current query. The plus button shows cross-references for the gene. If the results of a MaxLink search are visualized, the number of links to the query (MaxLink score) and the significance of the hit are shown in separate columns.
The Enriched terms box is shown below or next to the genes list. This contains two buttons to simplify analysis of pathway enrichment. The button Copy subnetwork genes will copy all genes in the subnetwork to the clipboard, and the Enrichment in PathwAX button will redirect the user to the PathwAX website, where the subnetwork genes can be pasted as a query into the search form.
Save
The save view allows you to download the query subnetwork either in XML format or as tab-separated values (TSV) file. TSV is a de facto standard for network data and can amongst other things be imported into Cytoscape. The different columns are: the confidence score, the FBS score, the gene pair, FBS scores for the 4 different coupling classes, LLRs for the different evidence types, LLRs for the species, and the class with strongest coupling. The links in the network file correspond to the strongest coupling class. For a comparative query the networks for the different species are given in separate TSV files.
Modify search
The modify search view brings back the current search and allows you to change the keywords or to review and modify the parameters.
FAQ
How do I cite FunCoup?
Please cite the latest paper from here if you are using the database or if you want to refer to the FunCoup algorithm. If you are using MaxLink please also cite Network-based Identification of Novel Cancer Genes (Östlund 2010).
How do I get only the couplings between my query genes
By default the FunCoup search returns the subnetwork of your query and the strongest coupled genes to your query. If you are only interested in links between your query genes, go into the advanced search options and set the "Expansion depth" on the "Sub-network selection" tab to 0.
Where can I get help?
See here
I know gene X and gene Y are coupled, why is there no link between them in FunCoup?
Have you tried lowering the confidence threshold?
It should be noted that the main objective of FunCoup is the prediction of
Why is there no network for species X?
Creating a functional coupling network for a species requires a lot of data including high-quality known couplings. The process involves a lot of manual work and is computationally demanding, we therfore focus on a small number of well studied model organisms.
Is the previous version of FunCoup still accessible?
Yes, all previous versions of the networks can be downloaded from the "Archive" page.
